Tutorial 5: L-R pairs analysis
The Nanostring CosMx Spatial Molecular Imaging (SMI) dataset used in this analysis was obtained from the official Nanostring repository: https://nanostring.com/products/cosmx-spatial-molecular-imager/nsclc-ffpe-dataset/. For L-R interaction analysis, we used a curated ligand-receptor pair list that combines entries from the SiGra framework and the IUPHAR/BPS Guide to PHARMACOLOGY database.
[7]:
import os
import torch
import pandas as pd
import scanpy as sc
from sklearn import metrics
from GraphST import GraphST
device = torch.device('cuda:1')
Define LR PAIR significance calculation
[ ]:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import ttest_rel
# this function is from scipy 1.11
def fdr(ps, *, axis=0, method='bh'):
# Input Validation and Special Cases
ps = np.asarray(ps)
ps_in_range = (np.issubdtype(ps.dtype, np.number)
and np.all(ps == np.clip(ps, 0, 1)))
if not ps_in_range:
raise ValueError("`ps` must include only numbers between 0 and 1.")
methods = {'bh', 'by'}
if method.lower() not in methods:
raise ValueError(f"Unrecognized `method` '{method}'."
f"Method must be one of {methods}.")
method = method.lower()
if axis is None:
axis = 0
ps = ps.ravel()
axis = np.asarray(axis)[()]
if not np.issubdtype(axis.dtype, np.integer) or axis.size != 1:
raise ValueError("`axis` must be an integer or `None`")
if ps.size <= 1 or ps.shape[axis] <= 1:
return ps[()]
ps = np.moveaxis(ps, axis, -1)
m = ps.shape[-1]
order = np.argsort(ps, axis=-1)
ps = np.take_along_axis(ps, order, axis=-1) # this copies ps
i = np.arange(1, m+1)
ps *= m / i
# Theorem 1.3 of [2]
if method == 'by':
ps *= np.sum(1 / i)
np.minimum.accumulate(ps[..., ::-1], out=ps[..., ::-1], axis=-1)
# Restore original order of axes and data
np.put_along_axis(ps, order, values=ps.copy(), axis=-1)
ps = np.moveaxis(ps, -1, axis)
return np.clip(ps, 0, 1)
load L-R pairs
[2]:
nano_gene = pd.read_csv('../Nanostring/all_in_one.csv')
gene_col = nano_gene.columns[-980:].to_list()
gene_col = [i.lower() for i in gene_col]
lr_pairs = pd.read_csv('../mergeall_LR_pairs_uniq.csv')
lr_pairs = lr_pairs.drop_duplicates()
print('Total number of pairs:', lr_pairs.shape[0])
lr_pairs['pair1'] = lr_pairs['Pair.Name'].apply(lambda x: x.split('_')[0].lower())
lr_pairs['pair2'] = lr_pairs['Pair.Name'].apply(lambda x: x.split('_')[1].lower())
print('Number of pair1', lr_pairs['pair1'].unique().shape[0])
print('Number of pair2', lr_pairs['pair2'].unique().shape[0])
# only keep those rows that both 'pair1' and 'pair2' in gene_col
lr_pairs = lr_pairs[lr_pairs['pair1'].isin(gene_col) & lr_pairs['pair2'].isin(gene_col)]
print('Number of pair1', lr_pairs['pair1'].unique().shape[0])
print('Number of pair2', lr_pairs['pair2'].unique().shape[0])
print('Total number of pairs:', lr_pairs.shape[0])
Total number of pairs: 2097
Number of pair1 693
Number of pair2 620
Number of pair1 189
Number of pair2 159
Total number of pairs: 480
[4]:
# label_map = {'Tumors': 0, 'Fibroblast': 1, 'Lymphocyte': 2, 'Mcell': 3, 'Neutrophil': 4, 'Endothelial': 5, 'Epithelial': 6, 'Mast': 7}
label_map = {'tumors': 0, 'fibroblast': 1, 'lymphocyte': 2, 'Mcell': 3, 'neutrophil': 4, 'endothelial': 5, 'epithelial': 6, 'mast': 7}
def load_Nano_data(id, root_path='./'):
data = pd.read_csv(os.path.join(root_path, 'all_in_one.csv'))
gene_feature_col = pd.read_csv(os.path.join(root_path, 'Lung9_Rep1_exprMat_file.csv')).columns[2:] #980 gene-related features
y = data[data['fov'] == id]['cell_type'] # the cell type is string type
y = y.map(label_map).to_numpy()
gene_fea = data[data['fov'] == id][gene_feature_col].to_numpy().astype(float)
return gene_fea, y, np.array([data[data['fov'] == id]['CenterY_local_px'], data[data['fov'] == id]['CenterX_local_px']]).T, gene_feature_col
[ ]:
from Utility.tools import load_train_data
import os
dataset = 'Nanostring'
id = '014'
pixel_size = 60
img_path = os.path.join('./Img_encoder/models/', dataset, id, 'img_emb.npy')
data_path = '../Data'
knn = 5
edge_index, fea, G, G_neg, gt = load_train_data(id=id,
knn=knn,
data_path=os.path.join(data_path, dataset),
img_path=img_path,
margin=pixel_size,
dataset=dataset)
Train the model
[ ]:
from Utility.train_w_data import train_
import yaml
raw_exp = []
enhanced_expression_matrix = []
cfg_path = './Best_cfg'
_raw_exp, gt, loc, gene_col = load_Nano_data(i, root_path='../Nanostring/')
raw_exp.append(_raw_exp)
gene_col = [i.lower() for i in gene_col]
raw_exp = np.concatenate(raw_exp, axis=0)
with open(os.path.join(cfg_path, dataset, id+'.yaml'), 'r') as f:
cfg = yaml.safe_load(f)
ari, label_pred, emb, enhanced_expression_matrix = train_(edge_index, fea, G, G_neg, gt, cfg)
enhanced_expression_matrix = np.concatenate(enhanced_expression_matrix, axis=0)
print(f'The final ARI is {ari}')
Training: 33%|███▎ | 489/1500 [00:18<00:38, 26.25it/s]
Reached the tolerance, early stop training at epoch 489
The final ARI is 0.9085177871448437
[9]:
# l2 norm
raw_exp = raw_exp / np.linalg.norm(raw_exp, axis=1, keepdims=True)
raw_expression_matrix = raw_exp
raw_expression_matrix = pd.DataFrame(raw_expression_matrix, columns=gene_col)
# enhanced_expression_matrix = np.where(enhanced_expression_matrix<0, 0, enhanced_expression_matrix)
enhanced_expression_matrix = enhanced_expression_matrix / (np.linalg.norm(enhanced_expression_matrix, axis=1, keepdims=True)+1e-10)
enhanced_expression_matrix = pd.DataFrame(enhanced_expression_matrix, columns=gene_col)
assert raw_expression_matrix.shape == enhanced_expression_matrix.shape
raw_expression_matrix = raw_expression_matrix.T
enhanced_expression_matrix = enhanced_expression_matrix.T
[ ]:
e_specific = []
r_specific = []
results = pd.DataFrame(columns=['LR_pair', 'p_value', 'p_value_enhanced'])
for id in range(len(lr_pairs)):
ligand_gene = lr_pairs.iloc[id]['pair1']
receptor_gene = lr_pairs.iloc[id]['pair2']
pair = lr_pairs.iloc[id]['Pair.Name']
# Extract expression data
ligand_raw = raw_expression_matrix.loc[ligand_gene]
receptor_raw = raw_expression_matrix.loc[receptor_gene]
ligand_enhanced = enhanced_expression_matrix.loc[ligand_gene]
receptor_enhanced = enhanced_expression_matrix.loc[receptor_gene]
# Perform paired t-test
test_result_raw = ttest_rel(ligand_raw, receptor_raw)
test_result_enhanced = ttest_rel(ligand_enhanced, receptor_enhanced)
results.loc[id] = [pair,test_result_raw[1],test_result_enhanced[1]]
# 对原始和增强 p 值进行 FDR 校正
results['FDR_raw'] = fdr(results['p_value'])
results['FDR_enhanced'] = fdr(results['p_value_enhanced'])
# 计算 -log10(FDR) 用于可视化
results['logFDR_raw'] = -np.log10(results['FDR_raw'] + 1e-10)
results['logFDR_enhanced'] = -np.log10(results['FDR_enhanced'] + 1e-10)
# 基于 FDR 判断显著性
results['Shared'] = (results['FDR_raw'] < 0.05) & (results['FDR_enhanced'] < 0.05)
results['Specific'] = (results['FDR_raw'] < 0.05) | (results['FDR_enhanced'] < 0.05)
results['Specific'] = results['Specific'] & (~results['Shared'])
results['Not Significant'] = (results['FDR_raw'] >= 0.05) & (results['FDR_enhanced'] >= 0.05)
# 将结果合并为 'Shared', 'Specific', 'Not significant' 三类
results['pair_type'] = results[['Shared', 'Specific', 'Not Significant']].idxmax(axis=1)
# 标记只在增强后显著的配体-受体对
results['specific_enhanced'] = (results['FDR_raw'] >= 0.05) & (results['FDR_enhanced'] < 0.05)
# 记录数量
e_specific.append(results['specific_enhanced'].sum())
r_specific.append(results['Specific'].sum() - results['specific_enhanced'].sum())
print(e_specific, r_specific)
[16] [12]
[ ]:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import ttest_rel
# this function is from scipy 1.11
def fdr(ps, *, axis=0, method='bh'):
# Input Validation and Special Cases
ps = np.asarray(ps)
ps_in_range = (np.issubdtype(ps.dtype, np.number)
and np.all(ps == np.clip(ps, 0, 1)))
if not ps_in_range:
raise ValueError("`ps` must include only numbers between 0 and 1.")
methods = {'bh', 'by'}
if method.lower() not in methods:
raise ValueError(f"Unrecognized `method` '{method}'."
f"Method must be one of {methods}.")
method = method.lower()
if axis is None:
axis = 0
ps = ps.ravel()
axis = np.asarray(axis)[()]
if not np.issubdtype(axis.dtype, np.integer) or axis.size != 1:
raise ValueError("`axis` must be an integer or `None`")
if ps.size <= 1 or ps.shape[axis] <= 1:
return ps[()]
ps = np.moveaxis(ps, axis, -1)
m = ps.shape[-1]
order = np.argsort(ps, axis=-1)
ps = np.take_along_axis(ps, order, axis=-1) # this copies ps
i = np.arange(1, m+1)
ps *= m / i
# Theorem 1.3 of [2]
if method == 'by':
ps *= np.sum(1 / i)
np.minimum.accumulate(ps[..., ::-1], out=ps[..., ::-1], axis=-1)
# Restore original order of axes and data
np.put_along_axis(ps, order, values=ps.copy(), axis=-1)
ps = np.moveaxis(ps, -1, axis)
return np.clip(ps, 0, 1)
[2]:
nano_gene = pd.read_csv('../Nanostring/all_in_one.csv')
gene_col = nano_gene.columns[-980:].to_list()
gene_col = [i.lower() for i in gene_col]
lr_pairs = pd.read_csv('../mergeall_LR_pairs_uniq.csv')
lr_pairs = lr_pairs.drop_duplicates()
print(lr_pairs.shape)
lr_pairs['pair1'] = lr_pairs['Pair.Name'].apply(lambda x: x.split('_')[0].lower())
lr_pairs['pair2'] = lr_pairs['Pair.Name'].apply(lambda x: x.split('_')[1].lower())
# only keep those rows can 'pair1' and 'pair2' in gene_col
lr_pairs = lr_pairs[lr_pairs['pair1'].isin(gene_col) & lr_pairs['pair2'].isin(gene_col)]
print(lr_pairs.shape)
(2097, 1)
(480, 3)
[3]:
# label_map = {'Tumors': 0, 'Fibroblast': 1, 'Lymphocyte': 2, 'Mcell': 3, 'Neutrophil': 4, 'Endothelial': 5, 'Epithelial': 6, 'Mast': 7}
label_map = {'tumors': 0, 'fibroblast': 1, 'lymphocyte': 2, 'Mcell': 3, 'neutrophil': 4, 'endothelial': 5, 'epithelial': 6, 'mast': 7}
def load_Nano_data(id, root_path='./'):
data = pd.read_csv(os.path.join(root_path, 'all_in_one.csv'))
gene_feature_col = pd.read_csv(os.path.join(root_path, 'Lung9_Rep1_exprMat_file.csv')).columns[2:] #980 gene-related features
y = data[data['fov'] == id]['cell_type'] # the cell type is string type
y = y.map(label_map).to_numpy()
gene_fea = data[data['fov'] == id][gene_feature_col].to_numpy().astype(float)
return gene_fea, y, np.array([data[data['fov'] == id]['CenterY_local_px'], data[data['fov'] == id]['CenterX_local_px']]).T, gene_feature_col
[12]:
import os
import numpy as np
path = './best_res/Nanostring/'
raw_exp = []
enhanced_expression_matrix = []
for i in range(1, 21):
_raw_exp, gt, loc, gene_col = load_Nano_data(i, root_path='../Nanostring/')
path_ = os.listdir(os.path.join(path, str(i).zfill(3)))[0]
_enhanced_expression_matrix = np.load(os.path.join(path, str(i).zfill(3), path_, 'recon_RNA.npy'), allow_pickle=True)
raw_exp.append(_raw_exp)
enhanced_expression_matrix.append(_enhanced_expression_matrix)
print(i)
gene_col = [i.lower() for i in gene_col]
raw_exp = np.concatenate(raw_exp, axis=0)
enhanced_expression_matrix = np.concatenate(enhanced_expression_matrix, axis=0)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
[15]:
# l2 norm
raw_exp = raw_exp / np.linalg.norm(raw_exp, axis=1, keepdims=True)
raw_expression_matrix = raw_exp
raw_expression_matrix = pd.DataFrame(raw_expression_matrix, columns=gene_col)
# enhanced_expression_matrix = np.where(enhanced_expression_matrix<0, 0, enhanced_expression_matrix)
enhanced_expression_matrix = enhanced_expression_matrix / (np.linalg.norm(enhanced_expression_matrix, axis=1, keepdims=True)+1e-10)
enhanced_expression_matrix = pd.DataFrame(enhanced_expression_matrix, columns=gene_col)
assert raw_expression_matrix.shape == enhanced_expression_matrix.shape
raw_expression_matrix = raw_expression_matrix.T
enhanced_expression_matrix = enhanced_expression_matrix.T
[28]:
e_specific = []
r_specific = []
results = pd.DataFrame(columns=['LR_pair', 'p_value', 'p_value_enhanced'])
for id in range(len(lr_pairs)):
ligand_gene = lr_pairs.iloc[id]['pair1']
receptor_gene = lr_pairs.iloc[id]['pair2']
pair = lr_pairs.iloc[id]['Pair.Name']
# Extract expression data
ligand_raw = raw_expression_matrix.loc[ligand_gene]
receptor_raw = raw_expression_matrix.loc[receptor_gene]
ligand_enhanced = enhanced_expression_matrix.loc[ligand_gene]
receptor_enhanced = enhanced_expression_matrix.loc[receptor_gene]
# Perform paired t-test
test_result_raw = ttest_rel(ligand_raw, receptor_raw)
test_result_enhanced = ttest_rel(ligand_enhanced, receptor_enhanced)
results.loc[id] = [pair,test_result_raw[1],test_result_enhanced[1]]
# 对原始和增强 p 值进行 FDR 校正
results['FDR_raw'] = fdr(results['p_value'])
results['FDR_enhanced'] = fdr(results['p_value_enhanced'])
# 计算 -log10(FDR) 用于可视化
results['logFDR_raw'] = -np.log10(results['FDR_raw'] + 1e-10)
results['logFDR_enhanced'] = -np.log10(results['FDR_enhanced'] + 1e-10)
# 基于 FDR 判断显著性
results['Shared'] = (results['FDR_raw'] < 0.05) & (results['FDR_enhanced'] < 0.05)
results['Specific'] = (results['FDR_raw'] < 0.05) | (results['FDR_enhanced'] < 0.05)
results['Specific'] = results['Specific'] & (~results['Shared'])
results['Not Significant'] = (results['FDR_raw'] >= 0.05) & (results['FDR_enhanced'] >= 0.05)
# 将结果合并为 'Shared', 'Specific', 'Not significant' 三类
results['pair_type'] = results[['Shared', 'Specific', 'Not Significant']].idxmax(axis=1)
# 标记只在增强后显著的配体-受体对
results['specific_enhanced'] = (results['FDR_raw'] >= 0.05) & (results['FDR_enhanced'] < 0.05)
# 记录数量
e_specific.append(results['specific_enhanced'].sum())
r_specific.append(results['Specific'].sum() - results['specific_enhanced'].sum())
print(e_specific, r_specific)
[21] [6]
[32]:
results.to_csv('figs/Nanostring/25-3-17/LR_pair_FDR.csv', index=False)
[ ]:
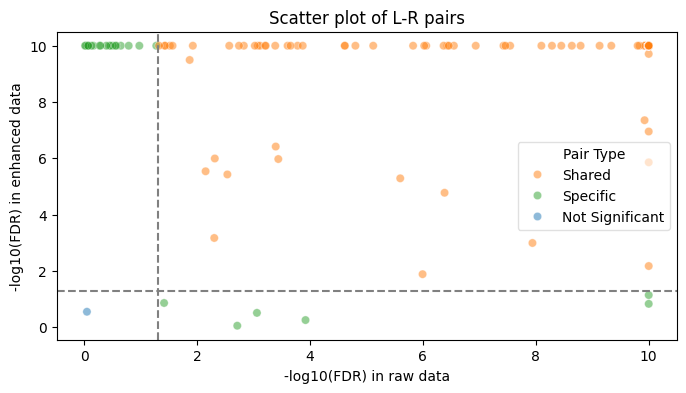
plt.figure(figsize=(8, 4))
from matplotlib import colormaps as cm
c_list = cm.get_cmap('tab10').colors
sns.scatterplot(data=results, x='logFDR_raw', y='logFDR_enhanced', hue='pair_type',
palette={'Shared': c_list[1], 'Specific': c_list[2], 'Not Significant': c_list[0]}, alpha=0.5, legend=False)
plt.legend(title='Pair Type', loc='center left', bbox_to_anchor=(1, 0.5))
plt.axhline(y=-np.log10(0.05), linestyle='--', color='grey')
plt.axvline(x=-np.log10(0.05), linestyle='--', color='grey')
plt.title("Scatter plot of L-R pairs")
plt.xlabel("-log10(FDR) in raw data")
plt.ylabel("-log10(FDR) in enhanced data")
## set legend to outside the plot
plt.legend(title='Pair Type', loc='best', framealpha=0.6)
# plt.legend(title='Pair Type', loc='center left', bbox_to_anchor=(1, 0.5))
plt.savefig('./figs/Nanostring/LR_pair_visualization.png', dpi=500)
plt.show()
[40]:
import pandas as pd
df = pd.read_csv('figs/Nanostring/25-3-17/LR_pair_FDR.csv')
df.head()
[40]:
| LR_pair | p_value | p_value_enhanced | FDR_raw | FDR_enhanced | logFDR_raw | logFDR_enhanced | Shared | Specific | Not Significant | pair_type | specific_enhanced | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ADM2_RAMP1 | 4.539310e-180 | 0.000000e+00 | 9.556442e-180 | 0.000000e+00 | 10.000000 | 10.0 | True | False | False | Shared | False |
| 1 | ANGPT1_ITGA5 | 0.000000e+00 | 0.000000e+00 | 0.000000e+00 | 0.000000e+00 | 10.000000 | 10.0 | True | False | False | Shared | False |
| 2 | ANGPT1_TEK | 5.220943e-01 | 1.642648e-97 | 5.332027e-01 | 1.886295e-97 | 0.273108 | 10.0 | False | True | False | Specific | True |
| 3 | ANGPT1_TIE1 | 2.302613e-30 | 0.000000e+00 | 3.131032e-30 | 0.000000e+00 | 10.000000 | 10.0 | True | False | False | Shared | False |
| 4 | ANGPT2_TEK | 4.105715e-37 | 1.349414e-156 | 5.745607e-37 | 1.623355e-156 | 10.000000 | 10.0 | True | False | False | Shared | False |
[41]:
import numpy as np
thred = -np.log10(0.05)
conditions = [
(df['logFDR_raw'] <= thred) & (df['logFDR_enhanced'] <= thred), # 左下
(df['logFDR_raw'] <= thred) & (df['logFDR_enhanced'] > thred), # 左上
(df['logFDR_raw'] > thred) & (df['logFDR_enhanced'] <= thred), # 右下
(df['logFDR_raw'] > thred) & (df['logFDR_enhanced'] > thred) # 右上
]
choices = ['bottom-left', 'top-left', 'bottom-right', 'top-right']
df['region'] = np.select(conditions, choices, default='unknown')
# 统计各区域数量
counts = df['region'].value_counts()
print(counts)
region
top-right 452
top-left 21
bottom-right 6
bottom-left 1
Name: count, dtype: int64